Table of Contents
Overview – Cystic Kidney Diseases
Cystic kidney diseases are a group of inherited disorders characterised by fluid-filled sacs (cysts) forming within the kidney. These cysts disrupt normal nephron architecture and function, leading to renal insufficiency. The two main types are Autosomal Dominant Polycystic Kidney Disease (ADPKD) and Autosomal Recessive Polycystic Kidney Disease (ARPKD), differing in inheritance, age of onset, and severity. These conditions can lead to end-stage renal disease and require lifelong monitoring and management.
Definition
- Renal cysts are dilated, filtrate-filled outpouchings of nephrons.
- Occur when nephrons fail to connect to collecting ducts → filtrate accumulates → cyst formation.
- May be isolated or part of a genetic syndrome.
Clinical Features (Shared)
- Abdominal or flank pain
- Haematuria (especially if cysts rupture)
- Urinary tract infections
- Renal insufficiency:
- Elevated serum creatinine
- Anaemia (↓ erythropoietin)
- Polyuria (impaired concentrating ability)
- Hypertension
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Aetiology
- Genetic inheritance: Autosomal dominant
- Prevalence: ~1 in 1000 (common)
Pathogenesis
- Tubules fail to connect to calyces → obstruction and cyst formation
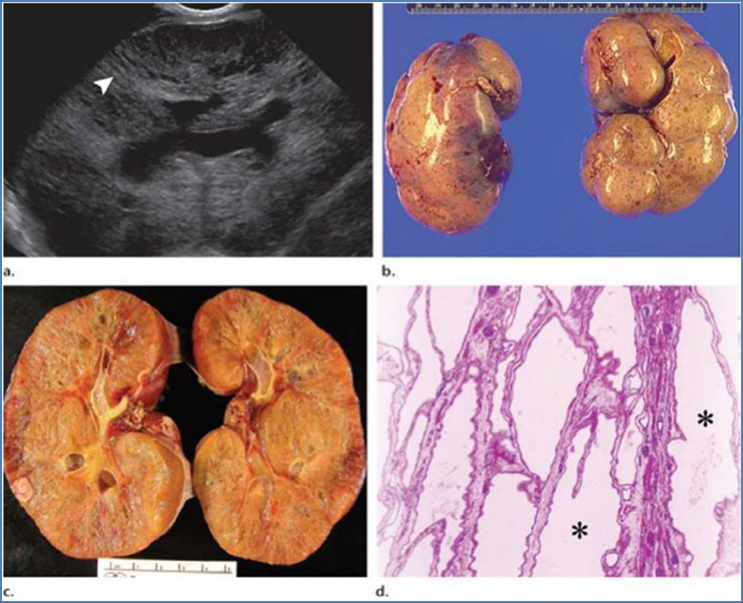
Morphology
- Bilateral enlarged kidneys
- Numerous cysts of varying size
- Some normal renal tissue remains
- Intermittent haemorrhage within cysts possible
Clinical Presentation
- Onset in adulthood (typically age 30–40)
- Symptoms:
- Flank or abdominal pain (from capsular stretching)
- Gross haematuria (cyst rupture)
- Hypertension and oedema
Complications
- Recurrent UTIs
- Progression to end-stage renal disease (around 50 years)
- Extra-renal features:
- Liver cysts (30%)
- Intracranial (berry) aneurysms (20%) → risk of subarachnoid haemorrhage
Management
- Supportive care
- Blood pressure control
- Dialysis or kidney transplantation for ESRD

Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Aetiology
- Genetic inheritance: Autosomal recessive
- Prevalence: Rare (~1 in 30,000 births)
Pathogenesis
- Nearly all tubules are cystic → severe and early-onset disease
Morphology
- Bilaterally enlarged kidneys with spongy appearance
- Uniform, small cysts
Clinical Presentation
- Detected in neonates or infants
- Bilateral abdominal masses (palpable kidneys)
- Poor urine concentrating ability
- Metabolic acidosis
- Hypertension
- Rapid progression to ESRD (within 15 years)
Prognosis
- 50% of neonates die in infancy
- Most survivors require renal replacement therapy before adolescence
Management
- Dialysis
- Kidney transplant
Summary – Cystic Kidney Diseases
Cystic kidney diseases, including ADPKD and ARPKD, are inherited disorders that lead to progressive kidney damage via cyst formation. ADPKD presents in adulthood with hypertension and renal insufficiency, while ARPKD manifests in infancy and carries a much poorer prognosis. Management focuses on symptom control, early detection of complications, and eventual renal replacement therapy. For a broader context, see our Renal Overview page.