Table of Contents
Overview – Thalassaemias
Thalassaemias are inherited haemoglobinopathies caused by reduced or absent synthesis of α- or β-globin chains, leading to ineffective erythropoiesis and chronic anaemia. Clinical severity varies from asymptomatic trait carriers to life-threatening transfusion-dependent disease. Complications arise from both the anaemia and the treatment — especially iron overload from chronic transfusions. Understanding the basic classification and pathophysiology of thalassaemias is essential for diagnosing and managing patients with microcytic anaemia.
Definition
- Thalassaemias are genetic disorders of haemoglobin synthesis
- Caused by defective production of either:
- Alpha chains (α-thalassaemia)
- Beta chains (β-thalassaemia)
- Leads to imbalanced globin chain production, resulting in ineffective erythropoiesis and chronic haemolysis
Aetiology
Alpha Thalassaemia
- Caused by deletion of 1 or more of 4 α-globin genes
- Severity increases with the number of deletions:
- 1 gene → silent carrier
- 2 genes → α-thalassaemia trait
- 3 genes → HbH disease
- 4 genes → hydrops fetalis (usually fatal in utero)
Beta Thalassaemia
- Due to point mutations in the β-globin gene
- Results in:
- β⁺ (partial deficiency)
- β⁰ (complete absence)
- Clinical forms:
- β-thalassaemia minor (trait)
- β-thalassaemia intermedia
- β-thalassaemia major (Cooley’s anaemia)
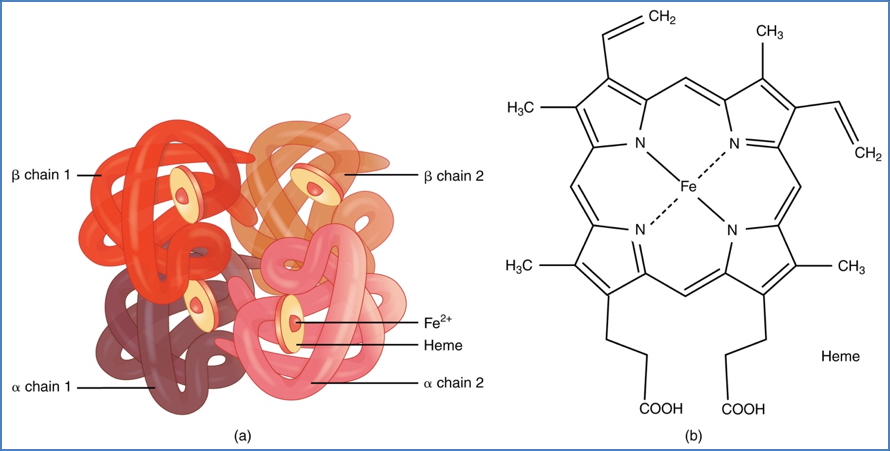
Pathophysiology
- ↓ Globin chain production → unmatched chains accumulate
- Unstable unmatched chains → RBC destruction in bone marrow and peripheral blood
- Ineffective erythropoiesis + haemolysis → chronic anaemia
- Bone marrow expands → skeletal deformities
- Compensatory mechanisms → hepatosplenomegaly
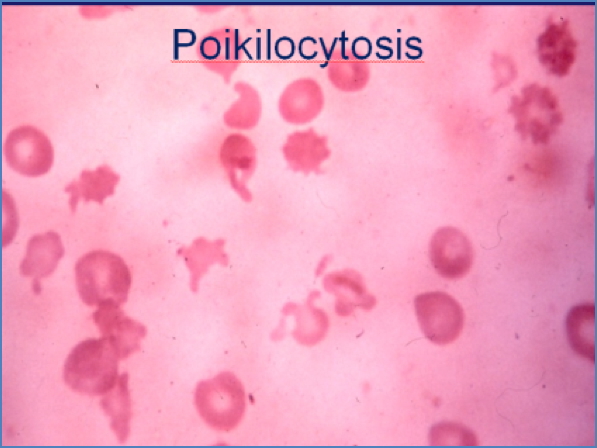
Microscopy
- Microcytic, hypochromic RBCs
- Anisopoikilocytosis (variable shape and size)
- Target cells, tear-drop cells, and basophilic stippling may be seen
Clinical Features
Mild Thalassaemia (Trait)
- Asymptomatic or mild anaemia
- Often detected incidentally
Moderate to Severe Disease
- Chronic fatigue, weakness
- Jaundice and dark urine (from haemolysis)
- Splenomegaly
- Delayed growth and puberty
- Skeletal changes: frontal bossing, maxillary overgrowth (due to marrow expansion)
Complications
- Iron overload:
- From transfusions and increased gut absorption
- Affects liver, heart, pancreas, pituitary
- Requires iron chelation therapy (e.g. deferoxamine)
- Infections (especially post-splenectomy)
- Osteoporosis and bone deformities
- Gallstones (from chronic haemolysis)
- Cardiomyopathy and heart failure (iron deposition)
Treatment
- Mild forms:
- No treatment required
- Genetic counselling recommended
- Moderate to severe forms:
- Regular blood transfusions to maintain Hb > 90–100 g/L
- Iron chelation therapy to prevent iron overload
- Splenectomy in selected cases
- Bone marrow transplantation: potential cure in severe cases
- Folic acid supplementation in chronic haemolysis
- Vaccinations (especially post-splenectomy): pneumococcus, meningococcus, Hib
Summary – Thalassaemias
Thalassaemias are inherited anaemias resulting from defective alpha or beta globin chain production. They range from silent carriers to transfusion-dependent disease with significant complications such as iron overload, bone deformities, and organ damage. Recognition and tailored management are crucial to improving quality of life. For a broader context, see our Blood & Haematology Overview page.