Table of Contents
Overview – Cystic Fibrosis
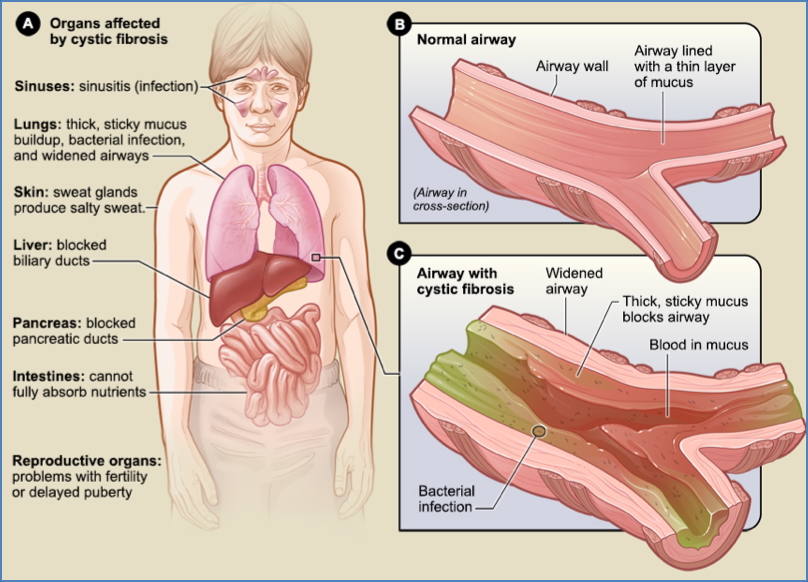
Cystic fibrosis is a common autosomal recessive genetic disorder caused by mutations in the CFTR gene, leading to thickened secretions that predominantly affect the lungs, pancreas, gastrointestinal tract, and sweat glands. This condition results in chronic infections, malabsorption, and infertility. With early diagnosis and emerging therapies like CFTR modulators, life expectancy and quality of life have improved dramatically for affected individuals.
Aetiology
- Genetic Cause:
- Autosomal recessive mutation of the CFTR gene on chromosome 7
- ~1 in 25 people are carriers in Caucasian populations
Pathogenesis
- CFTR Protein Dysfunction:
- Encodes chloride ion channels responsible for salt and water transport across epithelial cells
- Dysfunction results in dehydrated mucus and thickened exocrine secretions
- Multisystem Involvement:
- Lungs: Thick mucus → impaired mucociliary clearance → chronic infections
- Pancreas: Duct obstruction → enzyme deficiency → malabsorption
- Intestines: Malnutrition from poor nutrient absorption
- Reproductive tract: Vas deferens obstruction → male infertility
- Sweat glands: Excess chloride in sweat → hyponatraemia risk
Clinical Features
Respiratory
- Recurrent respiratory infections
- Persistent cough
- Crackles (crepitations) and rhonchi on auscultation
- Dyspnoea and wheezing
- Bronchiectasis and progressive lung damage
Pancreas
- Blocked pancreatic ducts → subclinical pancreatitis
- Pancreatic insufficiency → no enzyme delivery to duodenum
- → Malabsorption and poor growth
Gastrointestinal
- Steatorrhoea and malabsorption
- Failure to thrive and weight loss
- Fat-soluble vitamin deficiencies (A, D, E, K)
Reproductive
- Infertility in males due to congenital bilateral absence of vas deferens
Other
- Salty-tasting skin
- Electrolyte imbalances, especially hyponatraemia
Investigations
- Spirometry – Obstructive pattern (↓FEV1)
- Chest X-ray – Hyperinflation, gas trapping, bronchial wall thickening
- Sweat test (chloride concentration) – Historically diagnostic
- Genetic Testing – Definitive diagnosis (identification of CFTR mutations)
Management
Medical
- Enzyme replacement therapy – Creon Forte (pancreatic enzyme supplements)
- Salt supplementation – Salt tablets to prevent hyponatraemia
- Fat-soluble vitamins – ADEK supplementation
Respiratory
- Chest physiotherapy – Postural drainage, percussion to mobilise secretions
- Mucolytics – e.g. DNAse to reduce mucus viscosity
- Inhaled antibiotics – Tobramycin for recurrent Pseudomonas infections
- Bronchodilators – In selected cases
Other
- Lung transplant in end-stage disease
- Multidisciplinary team involvement (respiratory, gastroenterology, nutrition)
- CFTR modulators:
- Newer therapies targeting CFTR protein function
- Significantly reduce exacerbations and hospital admissions
Prognosis
- Median life expectancy has improved to ~40 years with optimal care
- Lung function deterioration is the most common cause of morbidity and mortality
Summary – Cystic Fibrosis
Cystic fibrosis is a life-limiting, autosomal recessive disorder caused by CFTR mutations, leading to multisystem involvement through thickened secretions. Early genetic diagnosis, aggressive respiratory and nutritional management, and the advent of CFTR modulators have dramatically improved patient outcomes. For broader context, visit our Respiratory Overview page or our Genetics & Cancer Overview page.