Table of Contents
Overview – Pharmacokinetics
Pharmacokinetics describes what the body does to a drug—from the moment it’s administered until it’s eliminated. It determines the time course of drug absorption, distribution, metabolism, and excretion (ADME), and ultimately controls the drug concentration at its site of action. For clinicians, understanding pharmacokinetics is essential to optimise dosing, avoid toxicity, and individualise therapy.
Definition
Pharmacokinetics is the study of the movement of drugs through the body, focusing on how they are absorbed, distributed, metabolised, and eliminated.
Factors That Influence Pharmacokinetics
Biological Membranes
Drugs must cross cellular membranes, which consist of:
- Bi-layered phospholipid structure
- Hydrophilic exterior, lipophilic interior
- Selective permeability
Drug Properties
- Lipid solubility → High solubility = high permeability
- Ionisation → Ionised drugs = poor permeability
- Determined by pKa (pH where drug is 50% ionised)
- Molecular size → Smaller = easier diffusion
Concentration Gradient
Most drugs use simple diffusion and move down their gradient until equilibrium is reached across compartments.
The Four Phases of Pharmacokinetics

1. Absorption
Definition
The process by which a drug moves from the site of administration into the plasma.
Key Points
- Rate of absorption affects intensity and duration of drug action.
- IV drugs bypass absorption — 100% bioavailability.
Mechanisms
- Simple diffusion (most drugs)
- Influenced by:
- pH and pKa
- Surface area
- Blood supply
- Tissue permeability
- Less commonly: facilitated diffusion, active transport.
Bioavailability (F)
- Percentage of active drug that reaches systemic circulation.
- IV = 100%
- Oral drugs may have low F due to poor absorption or first-pass metabolism.
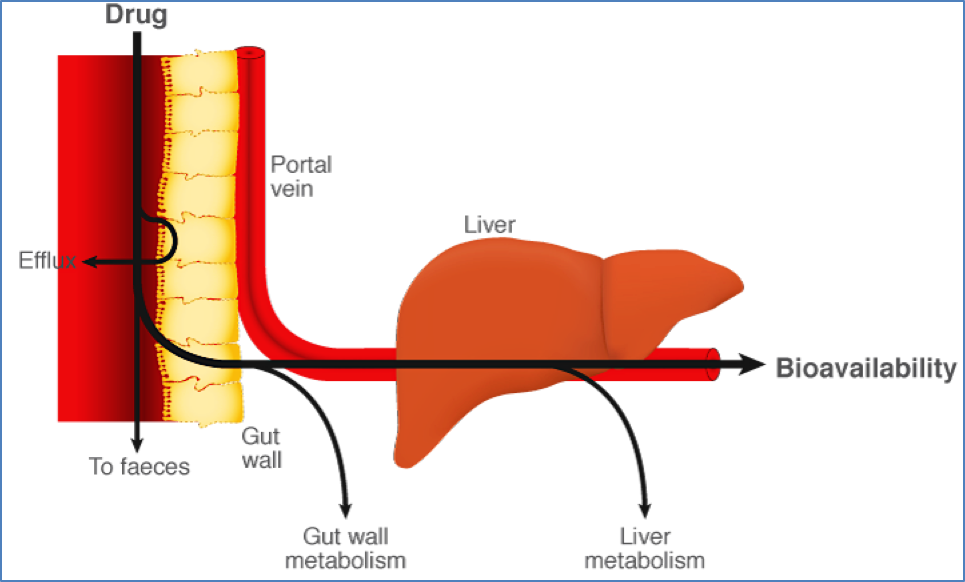
First-Pass Effect
- Drug is absorbed in GIT → Portal vein → Liver metabolises drug before it enters systemic circulation.
- ↓ Bioavailability
- Example: Lignocaine is effective subcutaneously but ineffective orally due to complete hepatic metabolism.
2. Distribution
Definition
Movement of the drug from plasma to various body compartments and the site of action.
Fluid Compartments
- Plasma
- Interstitial fluid
- Intracellular fluid
- Transcellular fluid (e.g. CSF, pleura)
- Fat
- Brain
Influencing Factors
- Lipid solubility: Lipophilic drugs cross membranes easily
- pKa and blood pH: Ionised drugs struggle to cross
- Protein binding: Only unbound drug can cross membranes
- Acidic drugs often bind albumin
- Regional blood flow: Determines tissue exposure
Volume of Distribution (Vd)
Volume of distribution (Vd) is a theoretical volume that describes how a drug is dispersed throughout the body relative to its concentration in the plasma.
Formula:
Vd = A / C
Where:
- A = Amount of drug administered (in mg)
- C = Plasma drug concentration (in mg/L)
Clinical Interpretation of Vd
- Vd reflects how extensively a drug distributes into body tissues versus staying in the plasma.
- It does not represent a real anatomical volume, but rather how the drug appears to distribute.
| Scenario | Interpretation |
|---|---|
| Vd ≈ Blood volume (~5 L) | Drug remains mostly in the plasma |
| Vd ≈ Total body water (~40–45 L) | Drug distributes uniformly in body fluids |
| Vd > Total body water (e.g. >50 L) | Drug is highly tissue-bound or fat-soluble |
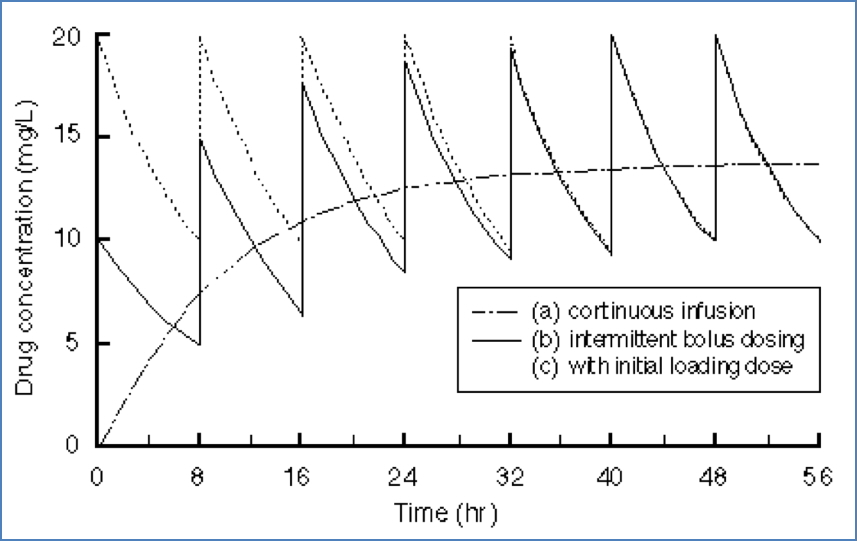
Clinical Application: Loading Dose
To quickly reach a desired steady-state concentration (Css), the loading dose (DL) can be calculated using Vd:
Formula:
DL = Vd × Css
Where:
- DL = Loading dose
- Css = Target steady-state concentration
Example: A lipophilic drug with a high Vd may require a larger loading dose to saturate peripheral tissues and achieve an effective plasma level.
Multicompartment Models
- Drugs distribute unevenly across tissues (e.g. fat vs muscle)
- Drugs redistribute from tissues → blood as plasma concentration decreases
Depots
- Tissues like fat can act as drug depots, storing lipophilic drugs (e.g. diazepam) for slow release
Barriers
- Some tissues limit drug entry:
- Placenta
- Blood–brain barrier
3. Metabolism (Biotransformation)
Definition
Chemical modification of drugs by the body, primarily in the liver.
Sites of Biotransformation
- Liver (main)
- GI tract
- Lungs
- Plasma
- Kidneys
Outcomes
- Prodrug activation → Inactive form becomes active
- E.g. Codeine → Morphine
- Conversion to active metabolite → Diazepam → Oxazepam
- Inactivation → Drug becomes pharmacologically inactive
- Toxic metabolite formation → Meperidine → Normeperidine
Factors Affecting Metabolism
- Drug interactions:
- Enzyme inhibitors ↑ toxicity (e.g. erythromycin + simvastatin)
- Enzyme inducers ↓ drug levels (e.g. phenobarbital + OCPs)
- Genetics:
- CYP2D6 absent in 7% of Caucasians
- Hyperactive in ~30% of East Africans
- Disease states: Liver/kidney/heart dysfunction impairs metabolism
- Hormones: Oestrogen can alter enzyme activity
- Age & gender: Enzyme expression varies with age and hormonal status
- Diet: Some foods, alcohol, vitamins alter enzyme activity
Metabolic Reactions
Phase I – Functionalisation
- Expose or modify functional groups
- Common reaction: Oxidation (mainly via Cytochrome P450 enzymes)

Phase II – Conjugation
- Add polar groups for water solubility → renal excretion
- Common conjugation types:
- Glucuronidation (most common)
- Sulfation
- Acetylation
- Methylation
- Glutathione conjugation

4. Elimination
Definition
The irreversible removal of drug from the body.
Routes of Elimination
- Kidneys (urine – main route)
- Lungs (e.g. anaesthetics, alcohol)
- Bile
- Faeces
- Saliva
- Breast milk
Renal Clearance Depends On:
- Glomerular filtration
- Tubular secretion
- Reabsorption
- Drug water solubility
Half-Life (t½)
- Time for plasma concentration to fall by 50%
- 5 × t½ = 97% elimination or steady state
Steady State (Css)
- Rate of drug input = rate of elimination
- Critical for maintenance dosing
Clearance (Cl)
- Volume of plasma cleared of drug per unit time (L/min)
- Calculated for whole body or specific organs
Elimination Kinetics
- First-order: Constant percentage per unit time (most drugs)
- Zero-order: Constant amount per unit time (e.g. high doses, saturation)

Clinical Applications of Pharmacokinetics
| Concept | Explanation |
|---|---|
| Loading dose | Large initial dose to reach therapeutic level quickly. Based on Vd. |
| Maintenance dose | Regular doses to maintain steady concentration. Based on clearance and t½. |
| Renal impairment | Requires dose adjustments to avoid toxicity. |
| Age/weight/comorbidity | Influence metabolism, distribution, clearance, and dosing needs. |
Summary – Pharmacokinetics
Pharmacokinetics explains how drugs are absorbed, distributed, metabolised, and eliminated from the body. It directly influences dosing, therapeutic effect, and toxicity risk. A strong grasp of ADME principles, metabolism pathways, and clearance kinetics is essential for safe and effective prescribing. For a broader context, see our Pharmacology & Toxicology Overview page.