Table of Contents
Overview – Retinoblastoma
Retinoblastoma is a malignant retinal tumour of early childhood, most often diagnosed before the age of five. It arises due to a mutation in the RB1 tumour suppressor gene, following the “two-hit hypothesis.” Retinoblastoma can occur unilaterally or bilaterally and presents with characteristic features such as leukocoria and strabismus. Early diagnosis and intraocular containment are essential, as prognosis drops significantly once the tumour becomes extraocular.
Definition
- Retinoblastoma is a malignant intraocular tumour of the retina, primarily affecting infants and young children.
- Can be unilateral or bilateral.
- High cure rates if detected early while the tumour remains intraocular.
Pathogenesis
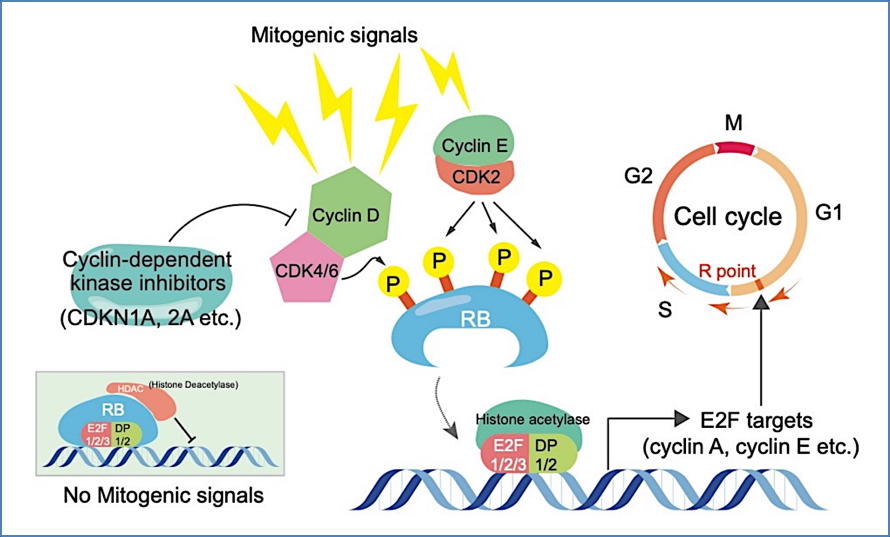
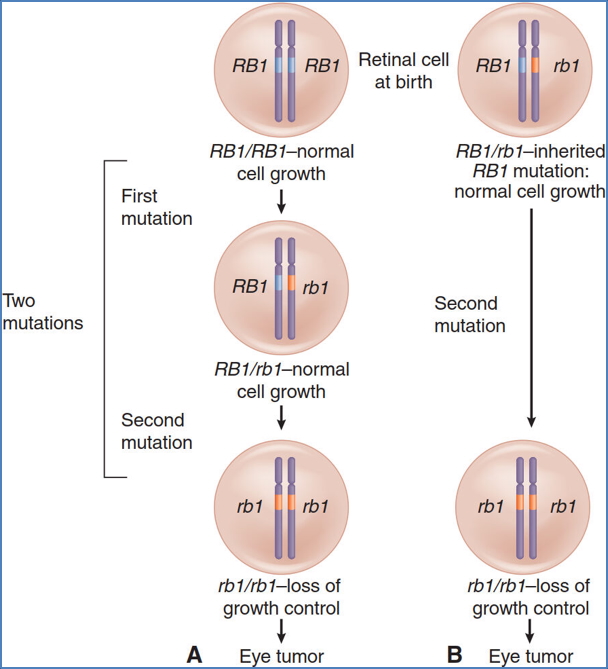
- Arises from biallelic inactivation of the RB1 tumour suppressor gene on chromosome 13.
- RB1 gene product (Rb protein) normally regulates cell cycle progression (G1 to S phase).
- Loss of both functional alleles leads to uncontrolled proliferation of retinoblasts.
- The “Two-Hit Hypothesis” applies:
- Hereditary form: First mutation is inherited (autosomal dominant); second mutation is acquired
- Sporadic form: Both mutations occur de novo
- Typically begins in foetal life, but becomes symptomatic in infancy or early childhood.
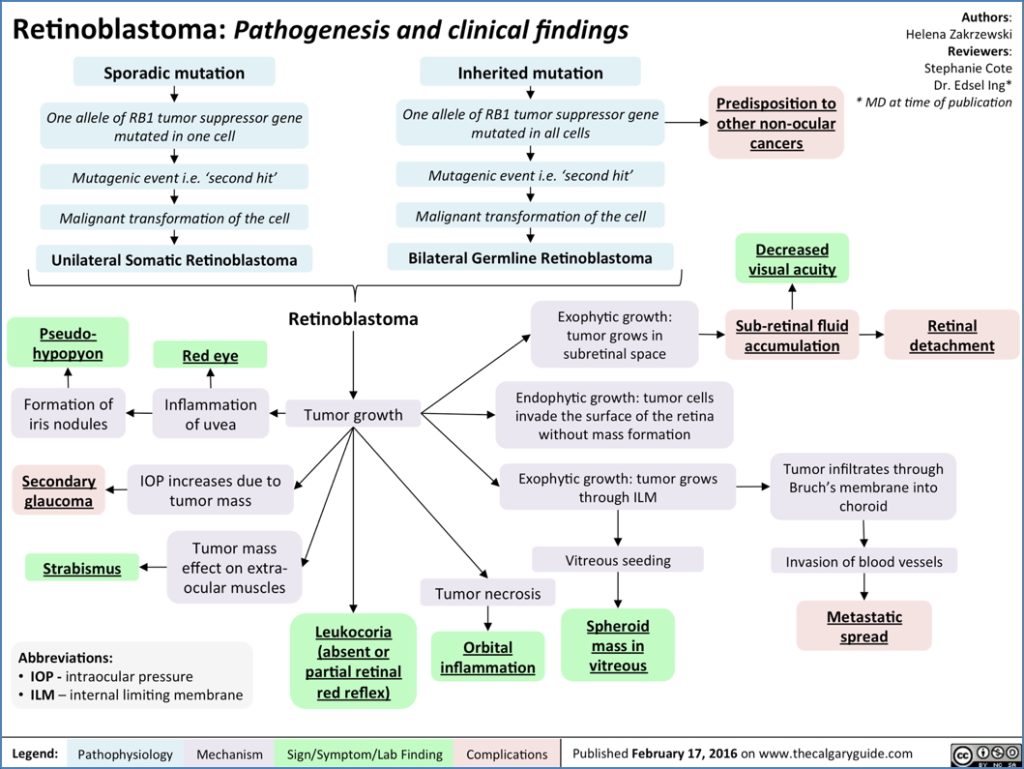
Clinical Features
- Leukocoria (white pupil reflex): Often first noticed in flash photography
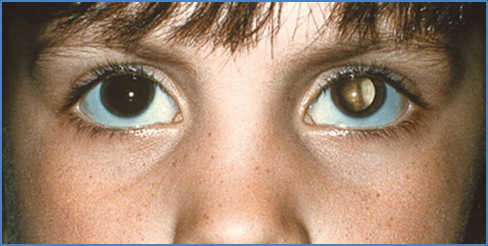
- Strabismus (misalignment of eyes): e.g. esotropia, exotropia
- Other signs:
- Visual impairment
- Eye pain
- Redness of the sclera
- Photophobia (light sensitivity)
- Unreactive pupil
- Symptoms usually evident before age 5.
Genetic Considerations
- RB1 gene mutation on chromosome 13
- Inheritance:
- 30% hereditary – autosomal dominant at the genetic level
- But autosomal recessive at the cellular level: both alleles must be inactivated
- Loss of heterozygosity is one way the second “hit” occurs

2. Int. J. Mol. Sci. 2020, 21(7), 2450; https://doi.org/10.3390/ijms21072450
- Hereditary cases are at increased risk of:
- Osteosarcoma
- Soft tissue sarcomas
- Melanoma
- Hence, regular cancer screening is advised for survivors.
Investigations
- Ophthalmoscopy (visualises intraocular tumour)
- Ultrasound of the eye – detects intraocular masses
- MRI or CT scan – assess for optic nerve invasion or extraocular spread
- Genetic testing – confirms RB1 mutation status
Management
Unilateral Retinoblastoma
- Enucleation (removal of the eye)
- Ocular prosthesis for cosmesis
Bilateral Retinoblastoma or Preserved Vision Cases
- Radiation therapy
- Laser photocoagulation or cryotherapy – for small tumours
- Systemic or intra-arterial chemotherapy – especially when there is a high risk of metastasis
- Close follow-up and monitoring are essential
Prognosis
- >90% 5-year survival if tumour is intraocular at diagnosis
- <10% 5-year survival if tumour is extraocular or invades the optic nerve
- Visual outcomes depend on laterality and treatment modality
Summary – Retinoblastoma
Retinoblastoma is a malignant eye tumour of childhood, driven by mutations in the RB1 gene and following the classic two-hit hypothesis. It presents with leukocoria and strabismus and may be unilateral or bilateral. Early intraocular diagnosis offers an excellent prognosis, but once the tumour extends beyond the globe, survival rates plummet. For a broader context, see our Genetics & Cancer Overview page.