Table of Contents
Overview – Neurofibromatosis
Neurofibromatosis is a genetic disorder characterised by the formation of benign tumours along nerves in the skin, brain, and other parts of the body. Neurofibromatosis is important to recognise due to its varied clinical presentations, potential complications, and its association with tumour-suppressor gene mutations.
Definition
Neurofibromatosis (NF) refers to a group of autosomal dominant genetic disorders that primarily affect nerve tissue, resulting in benign tumours called neurofibromas. These tumours can appear on or under the skin and can impact various organs and systems.
Aetiology
- Inheritance: Autosomal dominant
- Underlying mechanism: Mutation in tumour-suppressor genes (either NF1 or NF2)
- Genetic phenomenon: Illustrates pleiotropy — a single gene mutation causing multiple, seemingly unrelated phenotypic effects
Type 1 Neurofibromatosis (NF1)
- Mutation in the NF1 gene located on chromosome 17
- Encodes the neurofibromin protein, which normally regulates cell growth
- Affects approximately 1 in 3,000 individuals
Type 2 Neurofibromatosis (NF2)
- Mutation in the NF2 gene located on chromosome 22
- Encodes merlin (schwannomin), another tumour-suppressor protein
- Much rarer than NF1
Pathophysiology
- Both NF1 and NF2 are tumour-suppressor genes
- Disease develops when a second “hit” disables the remaining functional allele → loss of tumour suppression → uncontrolled cell growth
- This aligns with Knudson’s Two-Hit Hypothesis for tumour formation
Clinical Features
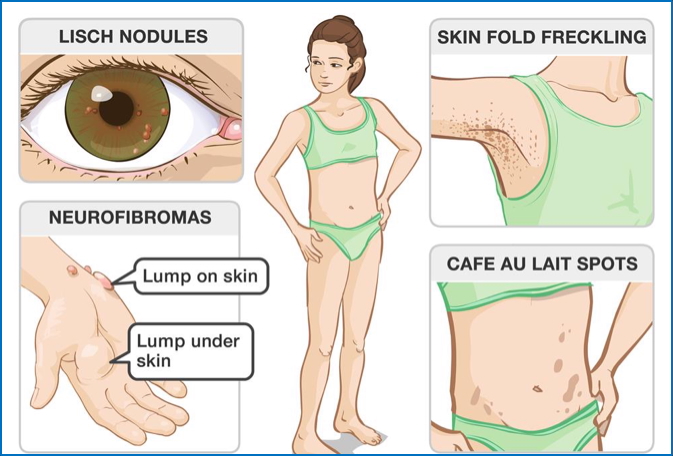
Type 1 NF (NF1):
- Cutaneous neurofibromas (lumps under the skin)
- Café-au-lait spots (flat pigmented skin lesions)
- Axillary or inguinal freckling
- Lisch nodules (iris hamartomas – benign eye growths)
- Bony abnormalities (e.g. scoliosis, bone dysplasia)
- Tumours in brain, spinal cord, or cranial nerves
- Learning disabilities and developmental delays
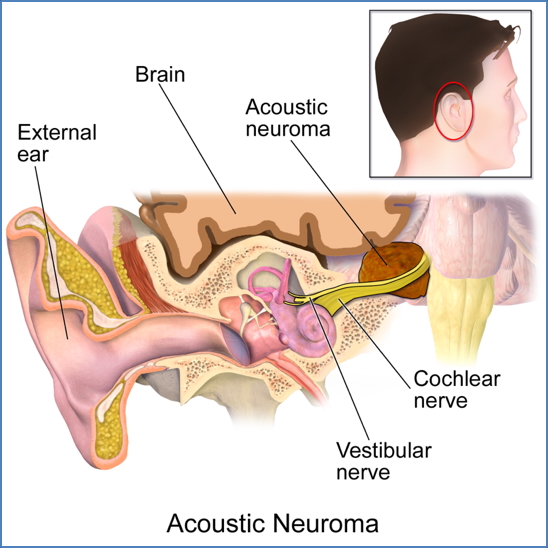
Type 2 NF (NF2):
- Bilateral vestibular schwannomas (tumours on auditory nerves)
→ tinnitus, hearing loss, balance problems (often in teenage years) - Additional cranial and spinal nerve schwannomas
- May present with cataracts or other visual disturbances
- Less likely to have skin findings seen in NF1
Investigations
- Clinical diagnosis supported by presence of key features (especially in NF1)
- MRI brain and spine – for nerve sheath tumours
- Audiometry – for suspected vestibular involvement (NF2)
- Genetic testing – confirmatory for both NF1 and NF2 mutations
Management
- Multidisciplinary approach (neurology, dermatology, oncology, ENT, ophthalmology)
- Regular surveillance for tumour progression
- Surgical excision of symptomatic neurofibromas or schwannomas
- Hearing support and interventions (e.g. cochlear implants for NF2)
- Genetic counselling for families
Complications
- Compression of surrounding nerves or tissues by tumours
- Malignant transformation of neurofibromas (especially in NF1)
- Hearing loss (NF2)
- Orthopaedic complications (scoliosis, bone deformities)
- Learning difficulties (NF1)
Differential Diagnosis
- Legius syndrome (similar café-au-lait macules without neurofibromas)
- Schwannomatosis (multiple schwannomas without vestibular involvement)
- Tuberous sclerosis
- Multiple endocrine neoplasia type 2B
Summary – Neurofibromatosis
Neurofibromatosis is a genetic disorder involving mutations in tumour-suppressor genes that lead to nerve-associated tumours and a variety of systemic manifestations. Type 1 and Type 2 have distinct clinical features and genetic mutations but share a common pathogenesis rooted in the two-hit hypothesis. Early identification and lifelong surveillance are essential for managing complications. For a broader context, see our Genetics & Cancer Overview page.