Table of Contents
Overview – Endocrine Pancreatic Tumours
Endocrine pancreatic tumours, also known as islet cell tumours, are a rare but clinically significant group of neoplasms arising from the hormone-producing cells of the pancreas. Although most are benign, their clinical relevance lies in their hormonal activity, which can lead to syndromes such as Zollinger-Ellison (gastrinoma) or hypoglycaemia (insulinoma). They may also occur as part of genetic syndromes like Multiple Endocrine Neoplasia (MEN). Understanding their diagnosis, hormonal effects, and treatment options is essential in both general and endocrine medicine.
Definition
Endocrine pancreatic tumours are neoplasms derived from islet cells that secrete peptide hormones. They are classified as functional (hormone-secreting) or non-functional tumours.
Aetiology
- Typically sporadic
- May be part of familial syndromes (e.g. MEN1, MEN2)
- Risk factors include genetic mutations in tumour suppressor genes (MEN1, RET)
Types & Pathogenesis
Gastrinoma (Zollinger-Ellison Syndrome)
- Pathogenesis: Gastrin-secreting tumour → hyperacidity → peptic ulcers
- Common site: Pancreas, duodenum, or stomach
Insulinoma
- Pathogenesis: Excessive, unregulated insulin production → hypoglycaemia
- Morphology: Usually small, solitary, and benign
Clinical Features
Gastrinoma
- Abdominal pain
- Dyspepsia
- Chronic diarrhoea (due to pancreatic enzyme inactivation)
Insulinoma
- Mild hypoglycaemia → anxiety, tachycardia, hunger, headache
- Severe hypoglycaemia → seizures, coma, possible death
Investigations
Gastrinoma
- Fasting serum gastrin
- Endoscopic ultrasound
- CT or MRI for localisation and staging
Insulinoma
- Fasting blood glucose + insulin levels
- CT/MRI abdomen
- Endoscopic ultrasound
Management
Gastrinoma
- Proton pump inhibitors (e.g. Omeprazole)
- Surgical resection (80% 5-year survival if resectable)
Insulinoma
- Surgical excision – curative in most cases
- Rarely malignant
General Management Options for Islet Cell Tumours
- Observation (if indolent or non-functioning)
- Hormone therapy (e.g. somatostatin analogues)
- Radiofrequency ablation / nuclear medicine therapy
- Radiation therapy (rarely needed)
Complications
- Gastrinoma → Peptic ulcer disease, malignancy
- Insulinoma → Recurrent severe hypoglycaemia
- Both may metastasise (especially gastrinomas)
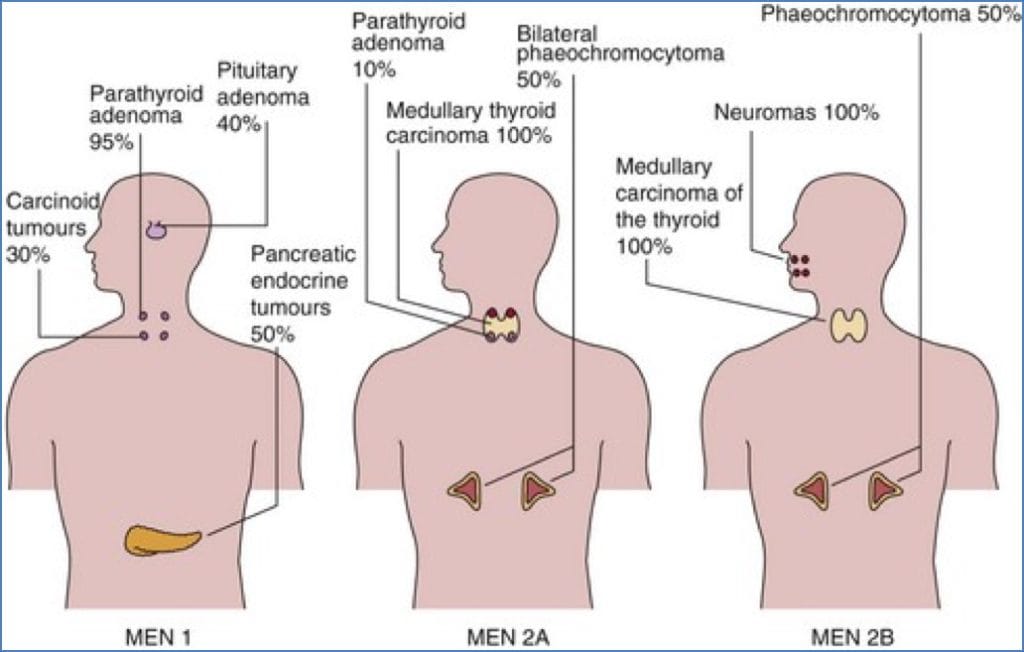
Syndromic Association: MEN Syndromes
MEN1 (Wermer’s Syndrome)
- Involves 2 or more of the following:
- Pituitary adenoma → visual field defects (e.g. bitemporal hemianopia)
- Pancreatic gastrinoma → peptic ulcer disease
- Pancreatic insulinoma → hypoglycaemia
- Parathyroid adenomas → hypercalcaemia from primary hyperparathyroidism
- Aetiology: Mutation in MEN1 tumour suppressor gene
MEN2 (Sipple’s Syndrome)
- Defined by:
- 100%: Medullary thyroid cancer
- 50%: Phaeochromocytoma
- 30%: Parathyroid hyperplasia/adenoma
- Aetiology: RET proto-oncogene mutation (MEN2A and MEN2B variants)
Summary – Endocrine Pancreatic Tumours
Endocrine pancreatic tumours, including gastrinomas and insulinomas, are rare hormone-secreting neoplasms with diverse clinical effects such as peptic ulcers or hypoglycaemia. Most are benign and surgically curable. They may occur sporadically or as part of MEN syndromes. For more context on related gastrointestinal conditions, see our Gastrointestinal Overview page.