Table of Contents
Overview – Sickle Cell Anaemia
Sickle cell anaemia is an inherited haemoglobinopathy caused by a structural mutation in the beta-globin gene. It leads to the formation of abnormal haemoglobin (HbS), which polymerises under low oxygen tension, distorting red blood cells into a sickle shape. These cells are rigid, poorly deformable, and prone to haemolysis and vascular occlusion. Sickle cell anaemia is most common in Afro-Caribbean populations and causes chronic anaemia, pain crises, and multiple systemic complications. Early diagnosis and preventive care are key to managing this lifelong condition.
Definition
- A genetic disorder of haemoglobin structure
- Characterised by production of HbS instead of normal HbA
- Leads to sickle-shaped red cells, chronic haemolysis, and vaso-occlusive complications
Aetiology
- Autosomal recessive inheritance
- Point mutation in β-globin gene (Glu → Val substitution)
- Prevalent in African, Afro-Caribbean, and Mediterranean populations
Pathophysiology
- Under low oxygen tension, HbS becomes insoluble
- Polymerisation of HbS → RBCs assume sickle shape
- Sickle cells are:
- Rigid
- Sticky
- Poorly deformable
- Consequences:
- Capillary obstruction → local ischaemia and infarction
- Haemolysis → chronic anaemia and elevated bilirubin
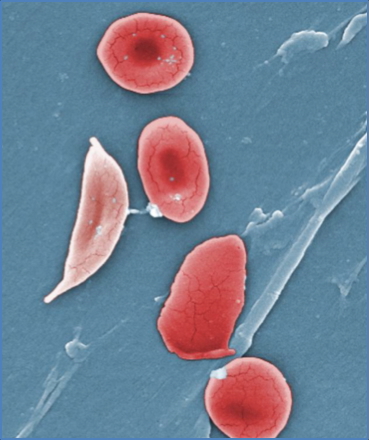
Microscopy
- Sickle-shaped red cells on peripheral smear
- Target cells and nucleated RBCs may also be seen
- Evidence of chronic haemolysis (e.g. ↑ reticulocytes)
Clinical Features
Anaemia Symptoms
- Fatigue
- Pallor
- Exertional dyspnoea
Vaso-Occlusive Symptoms
- Pain crises (most characteristic feature)
- Sudden, severe pain due to ischaemia
- Typically affects chest, abdomen, bones, and joints
- May occur infrequently or multiple times per year
- Dactylitis: swelling of hands and feet (common in infants)
- Frequent infections (due to functional asplenia)
- Delayed growth and puberty
Treatment
- No definitive cure (except potential stem cell transplant)
- Hydroxyurea: increases HbF and reduces pain crisis frequency
- L-glutamine: reduces frequency and severity of crises
- Analgesia during pain episodes
- Vaccination and antibiotic prophylaxis (e.g. penicillin) to prevent infections
- Folic acid supplementation for red cell turnover
- Blood transfusions in selected cases (e.g. stroke prevention)
- Bone marrow transplant: potentially curative in children
Complications
- Stroke
- Pulmonary hypertension
- Heart failure
- Renal disease
- Retinopathy and blindness
- Leg ulcers
- Avascular necrosis of bone
- Gallstones (from chronic haemolysis)
- Priapism
- Delayed growth and puberty
- Recurrent miscarriage or complications in pregnancy
Summary – Sickle Cell Anaemia
Sickle cell anaemia is an inherited disorder leading to sickling of red cells under low oxygen, causing haemolysis and painful vaso-occlusive crises. It is common in individuals of Afro-Caribbean descent and presents with anaemia, recurrent pain episodes, and systemic complications. Management focuses on crisis prevention, symptom relief, and long-term organ protection. For a broader context, see our Blood & Haematology Overview page.